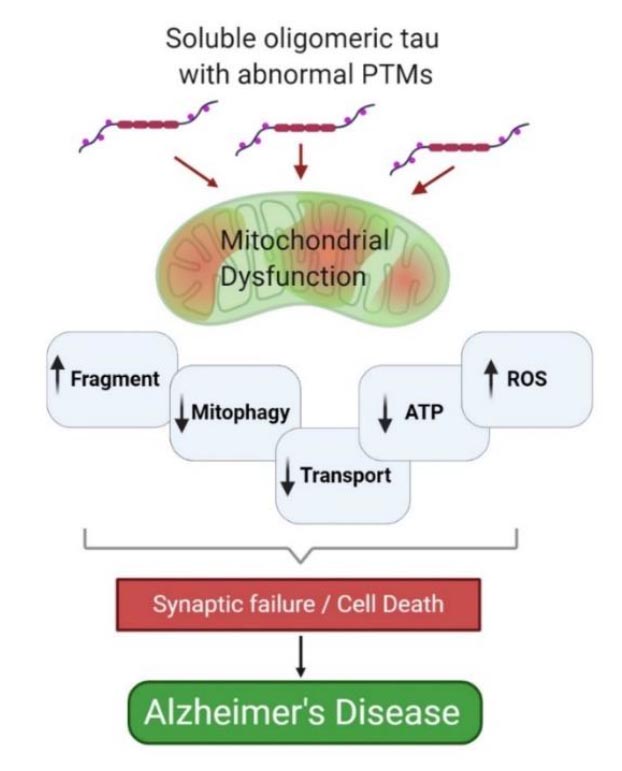
Tau is a central player in the pathogenesis of numerous age-related neurodegenerative diseases, with Alzheimer’s disease (AD) being the best example. Tau from AD brain is defined by aberrant posttranslational modifications (PTMs), including increases in phosphorylation and acetylation at specific epitopes. The UNDERLYING PREMISE of this proposal is that specific, disease relevant PTMs impair tau function which negatively impacts neuronal health. While the formation of insoluble fibrillary structures is influenced by PTMs, data strongly indicate that soluble forms of abnormally modified tau are the mediators of neuronal toxicity. There is data indicating that overexpression of AD-relevant forms of tau results in increased levels of fragmented, dysfunctional mitochondria, which may be due to in part due to impaired mitophagy, as well as other perturbations of mitochondrial quality control (MQC) mechanisms. However, a CRITICAL KNOWLEDGE GAP is how these modified tau species, when present at physiologically relevant levels, influence mitochondrial and neuronal health. The OVERALL HYPOTHESIS of this proposal is that tau with AD-relevant PTMs exerts toxic effects through impairing MQC mechanisms. An impaired ability to resolve mitochondrial stress would increase the presence of less functional mitochondria with a concomitant increase in oxidative stress and neuronal dysfunction. The overall result would be an earlier onset of an aged neuron phenotype. The NOVELTY of this project stems in part from its use of single-copy transgenic tau models that avoid overexpression, as well as the inclusion of age as a variable in a genetic model organism. The nematode C. elegans benefits from a vast repertoire of genetic, transgenic and genomic resources that will be leveraged to investigate the molecular underpinnings of AD and to define the precise mechanism through which tau PTMs compromise mitochondrial function and accelerate neuronal aging. Our preliminary data support this approach as worms with single copy expression of tau with specific AD-relevant PTMs in mechanosensory neurons show a significant increase in age-dependent neurodegeneration and a suppression of stress-induced mitophagy. These and other preliminary data provide a strong foundation for the studies in this application. The aims of this proposal are: (1) To determine the impact of AD relevant tau PTMs on mitochondrial stress responses and how this influences healthy aging of neurons, (2) To test the hypothesis that tau with AD relevant PTMs impairs mitochondrial dynamics and mitophagy, and (3) To address whether enhancing MQC is a viable therapeutic avenue. The relative contribution of these responses to neuronal age-dependent deficits will be tested using unique genetic resources available in worms. The IMPACT of these studies will be to provide crucial new insights into the mechanisms by which pathological tau species compromise mitochondrial function and neuronal health.
Tau Post-Translational Modifications and Mitochondrial Quality Control
 Dept. of Medicine, Nephrology Division at the University of Rochester Medical Center
Dept. of Medicine, Nephrology Division at the University of Rochester Medical Center
- Golisano Children’s Hospital Celebrates 10 Years of Changing Lives July 17, 2025
- EIOH Receives $2.1 Million to Expand Training and Public Health Initiatives July 16, 2025
- Burnout, Mental Health & Self-Compassion: Lessons from Medicine for All of Us July 16, 2025
- Can Type 1 Diabetes Be Cured? Diabetes Myths Debunked July 15, 2025
- University of Rochester Awarded Prestigious Aging Research Center Grant July 15, 2025
- Renovated EIOH Clinic at School 17 Expands Access to Care for Rochester Families July 15, 2025
- Remembering Ray Mayewski: An Innovative CMO and a Doctors’ Doctor July 14, 2025
- How to Prevent Brain Risks Linked to Post-Surgery Confusion July 9, 2025
- How to Manage Anxiety July 9, 2025
- New Eastman Dental Foundation Board President and Members July 8, 2025
There are no comments
Add yours